From Sequence to Structure: The Protein Folding Enigma
– 6 min read
By Aryan Boruah
Introduction
Proteins are basic building blocks of life and essential to all life forms. They are amazing little nano-biomachines and believe me they are incredible. Every function in our body depends on proteins. Now imagine a world where the secrets of life’s building blocks are laid bare, where the complex dance of molecules can be predicted with astonishing accuracy—the protein folding problem—a puzzle that has captivated scientists for decades. Proteins, the workhorses of cells, perform countless functions vital to life. But before they can do their job, these long chains of amino acids must fold into precise three-dimensional shapes.
Problem Statement
Despite significant advancements in the understanding of molecular structural biology, the protein folding problem remains one of the most daunting puzzle to solve. A protein is made of a string of multiple iterations of 20 varieties of amino acids. Now the key thing is, the structure is somehow specified by the amino acid sequence. Simply we can say that proteins are specified by their genetic sequence and then they fold up into a 3D structure. Now here comes the hard part we want to know what the 3D structure is because the 3D structure will determine its function. This indicates that the structure can be mapped to the function. And this in essence the protein folding problem “Can we just from the amino acid sequence (the 1-dimensional string of letters) immediately computationally predict the 3D structure?
Challenge
The protein folding problem always reminds me of Cyrus Levinthal. Cyrus Levinthal gave the Levinthal Paradox which states that “Finding the native folded state of a protein by a random search among all possible configurations can take an enormously long time. Yet proteins can fold in seconds or less”. In 1969, Levinthal noted that because of a very high degree of freedom in an unfolded polypeptide chain, the molecule has an astronomical number of possible conformations. Cyrus Levinthal estimated that a typical protein could fold in more than 10^140 ways. Well don’t take this number too seriously though, the number of possible folding actually depends on the size of the protein. Small proteins may have as few as 10^50 while some large ones can have mind-blowing 10^300 possible folding.
Now here comes the major challenge to predict the possible stable conformations of the protein out of those 10^300 conformations by looking at the chain of amino acid sequence.
Findings and solutions
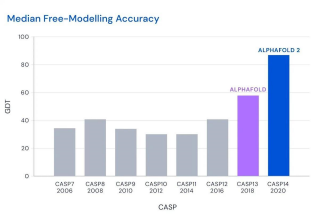
Initially it was believed that the protein folding problem is extremely hard and practically impossible to solve. It is thought of as equivalent to Fermat’s Last Theorem, but for biology. Amazingly, Google’s parent company Deepmind came up with a solution to this problem, the AlphaFold system. The AlphaFold system is the first non-experimental method that achieved accuracy comparable to experimental methods, and the second iteration of the AlphaFold system, AlphaFold2 released in July 2021 solved the 50- year-old grand challenge in biology. “Solved” here it means that these computational methods were able to achieve better predictions with high GDT (Global Distance Test) on CASP competition. To give a little context, CASP is Critical Assessment of Protein Structure prediction, is a community-wide, worldwide experiment for protein structure prediction taking place every two years since 1994. In CASP13, 2018 AlphaFold scored 58 GDT scores on the hardest class of proteins, and in 2021, AlphaFold2 scored a whooping 87 which is a huge improvement.
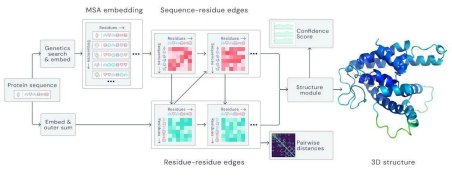
The high accuracy of AlphaFold2 lies in its backend working. A folded protein can be conceptualized as a "Spatial Graph," where the nodes represent residues and the edges link residues that are in close proximity. The graph obtained is important for the understanding of the how the amino acid residues interact within protein as well as their evolutionary history. The AlphaFold2 does the same, it fundamentally works on an attention based neural network system, trained end to end, that attempts to interpret the structure of the spatial graph, while simultaneously reasoning over the implicit graph it constructs. AlphaFold2 leverages the evolutionarily related sequences, multiple sequences alignment to find homogeneity in amino acid sequences across species, and a representation of amino acid pairs to refine the graph using a pair representation matrix. By iterating this process many times, it develops strong predictions of the underlying physical structure of
the protein. The AlphaFold2 system can be broken down into 2 blocks: Evoformer and Structure module
Evoformer Block
The core of AlphaFold2 is the Evoformer, a deep neural network block designed to process and integrate information from the MSAs and pair representations.
As I mentioned before, AlphaFold2 uses an attention-based mechanism that happens extensively within the Evoformer. This allows the model to focus on relevant parts of the sequence and spatial information, dynamically adjusting its attention based on the context provided by the MSA and pair representations.
Then interestingly, the Evoformer iteratively refines the MSA and pair representations. Through multiple layers and passes, the network progressively improves the quality of its representations, capturing increasingly detailed and accurate structural information.
Structure Module
After the Evoformer processes the input data, the refined pair representation is fed into the Structure Module. This module interprets the spatial relationships encoded in the pair representation to predict the final 3D coordinates of the protein atoms.
Summarizing this mechanism:
The system has an input of amino acid sequence and MSA, the Evoformer Block processes, and integrates evolutionary and spatial information using attention mechanisms. Then the iterative refinement happens within the Evoformer that enhances representations through multiple layers. The Structure Module interprets refined representations to predict 3D coordinates. Then it utilizes a graph-based approach to model residue interactions to provide reliability scores for the predicted structures.
Opinion and conclusion
Despite have ~90 GDP, critics have pointed out that it is still a failure and some of the most interesting cases are the ones that AlphaFold didnot performed well such complexes of proteins called oligomers in which several amino acids are interacting. Also, there is a general problem with artificial intelligence, that they only learn to extract patterns from data in
which they have been trained. This means the data has to exist in the first place. So, if there is an entirely new function that doesn't make appearance in the dataset, it may remain undiscovered. To conclude, the AlphaFold2 success won't be the end of the story. Much advancements are required and of course, we need data to train artificial intelligence in the first place. Still, it's a remarkable achievement and in the future protein folding prediction by artificial intelligence will cut huge expenses and time. Protein structure prediction can have a huge impact on protein engineering, this would help researchers to develop proteins that stimulate the immune system to fight cancer, universal flu vaccine, or proteins that break down microplastics.
Reference:
1) Image: https://deepmind.google/discover/blog/alphafold-a-solution-to-a-50-year-old grand-challenge-in-biology/ , First page protein structure
2) What Is AlphaFold? | NEJM (youtube.com)
3) AlphaFold informative video (Youtube)