A Turning Point in Huntington’s Disease Treatment: AMT-130 Gene Therapy
Novel gene therapy AMT-130 has completed its phase I/II clinical trials and could be the answer to the long-standing search for a Huntington’s disease treatment.
For individuals who inherit the Huntington’s disease mutation, certainty comes without remedy: a future shaped by progressive neurological decline with no means of altering its course. Affecting approximately 1 in 10,000 people worldwide, Huntington’s disease has been a stark example of how a known genetic cause does not necessarily translate into a modifiable outcome. Recent advances in molecular biology, however, are beginning to challenge the idea that genetic predisposition is immutable.
Huntington’s disease is a genetic neurodegenerative disorder with a devastating clinical course, characterised by gradual deterioration in cognition, impaired motor control, and psychiatric symptoms including anxiety, depression, and psychosis. First documented in 1872, the disease has been extensively studied, yet there is still a lack of effective treatment. Huntington’s is caused by a genetic mutation, causing a repeat expansion within the huntingtin gene. In a healthy person, there are between 10 and 35 CAG repeats in the huntingtin gene, whereas a Huntington’s patient will inherit over 35 repeats. These repeats result in a larger, toxic form of the huntingtin protein being produced within neurons, which builds up and prevents normal neuronal functioning. Huntington’s patients often experience symptom onset later in life, around 40 years old, after which symptoms become progressively worse, and individuals may live for a further ten to thirty years (McColgan and Tabrizi, 2017). This debilitating clinical course highlights the dire need for a treatment that directly targets the underlying genetic cause and actively slows disease progression, as only symptomatic treatment is available.
Gene therapy has emerged as a transformative approach in modern medicine, moving beyond conventional drug design, which typically targets proteins, and intervening directly at the genetic level. By delivering carefully engineered genetic material into specific cells, gene therapies can replace faulty genes, silence harmful ones, or introduce protective molecules. This strategy has already transformed treatment landscapes for multiple inherited diseases, and many researchers are working towards the use of gene therapy in Huntington’s disease. The recent breakthrough in AMT-130, a novel gene therapy targeted to treat Huntington’s disease, provides a hopeful indication that those who inherit the Huntington’s disease gene may not have to face the devastating symptoms. AMT-130 is delivered directly into key brain regions through a neurosurgical procedure guided by MRI imaging. The gene therapy uses Adeno-associated virus (AAV) as a delivery vehicle, which is harmless and very effective at entering human cells, neurons in this case, and unloading their genetic cargo. A specific genetic code has been inserted into the AAV genome, which, when expressed, produces a small RNA (microRNA) molecule. This microRNA recognises and binds to the messenger RNA responsible for synthesising the toxic huntingtin protein (Evers and Pavlina Konstantinova, 2020). By blocking this messenger RNA, the cell machinery can no longer manufacture the huntingtin protein, reducing its accumulation over time and the consequent symptoms. AMT-130 remains in neurons after treatment and binds to a specific protein, which protects it from our cells’ defences (“Artificial miRNA slows Huntington’s”, 2025). Thus, AMT-130 will continue to express the therapeutic microRNA for years, and potentially for the duration of the patient’s life.
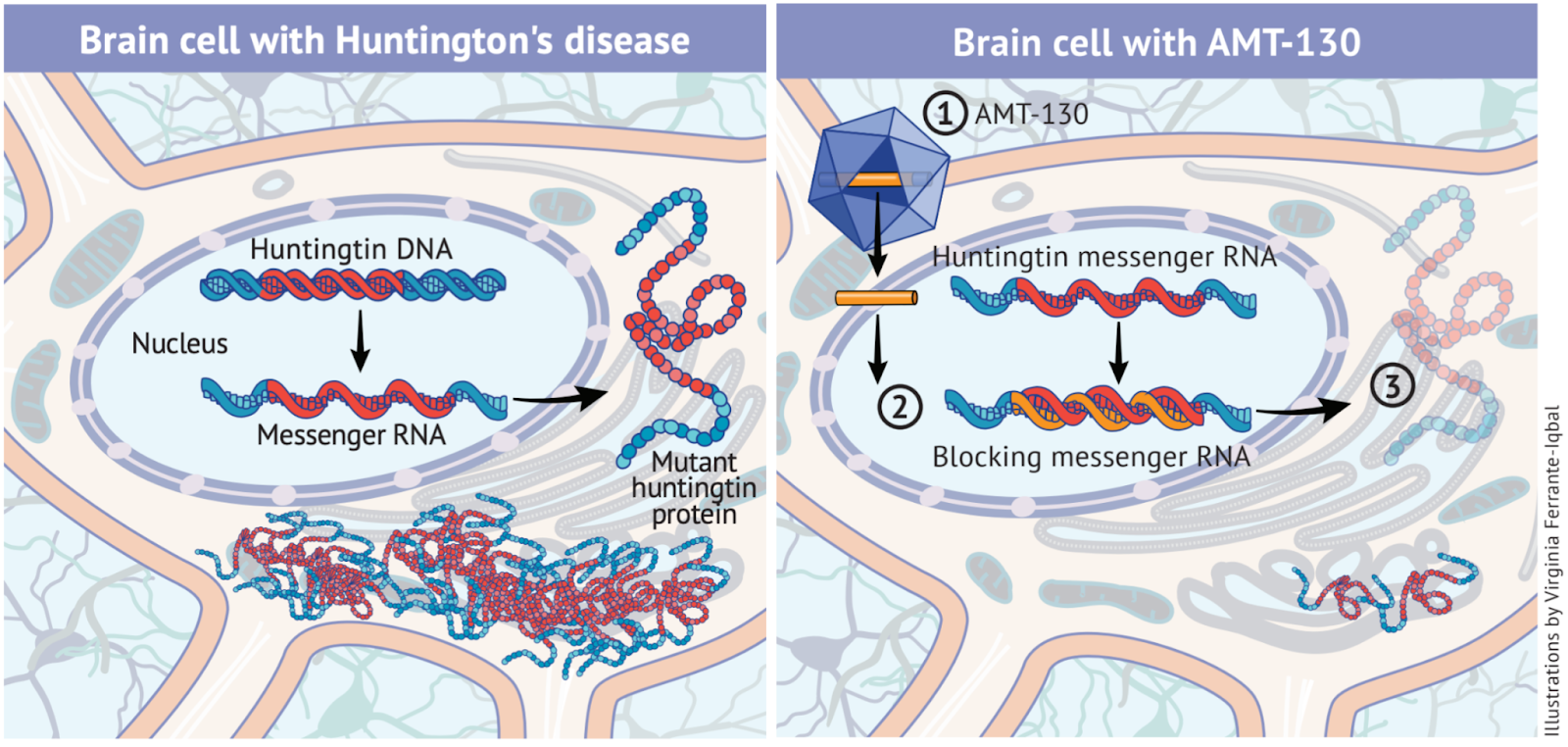
Figure 1. Mechanism of mutant huntingtin production and its suppression by AMT-130, adapted from (uniQure, n.d.).
In a neuron affected by HD (left), the mutant HTT gene is transcribed into messenger RNA (mRNA), which is exported to the cytoplasm and translated into mutant huntingtin protein that accumulates and disrupts cellular function. AMT-130 treatment (right) introduces a therapeutic microRNA which binds to mutant huntingtin mRNA, preventing its translation and thereby reducing production of the toxic huntingtin protein and its pathological effects.
This promising drug has recently completed its phase I/II clinical trials, meaning it has been tested in Huntington’s patients in order to establish its safety, dosage, and efficacy. These trials involved 29 patients who had been diagnosed within the last few years and were suffering from early symptoms. Of these patients, 17 received a high dose of AMT-130 and 12 received a low dose. The results of this clinical trial are extremely encouraging, demonstrating a 75% slowing of disease progression across the 36-month follow-up period, measured through assessments of motor performance, cognitive function, and functional independence. Further measures, such as a 60% reduction in Total Functional Capacity decline (an indicator of an individual’s ability to carry out everyday tasks and live independently) reinforced the therapy’s efficacy. Importantly, a neuronal damage biomarker called neurofilament light chain (NfL) was reduced to below baseline levels, indicating a significant decline in neuronal damage. The gene therapy was also well tolerated throughout the trial, with no serious adverse effects reported (uniQure N.V., 2025). Collectively, these outcomes suggest that AMT-130 can meaningfully alter the course of Huntington’s disease, providing hope for a healthier and autonomous life for those affected by the disease.
The next stage in its progress lies with the Food and Drug Administration (FDA), the body responsible for approving pharmaceuticals for clinical use. AMT-130 has been granted Breakthrough Therapy Designation, enabling closer collaboration between the researchers and the FDA to accelerate the assessment of safety and efficacy (uniQure, 2025). The early success of AMT-130 presents profound implications for the future of Huntington’s disease research and treatment, possibly shifting the disease from an inevitably debilitating condition to a manageable one. If long-term follow-up continues to demonstrate a therapeutic effect, AMT-130 could go on to establish a blueprint for future gene therapy designs targeting other inherited neurodegenerative disorders.
Evers, M.M. and Pavlina Konstantinova (2020). AAV5-miHTT gene therapy for Huntington disease: lowering both huntingtins. Expert Opinion on Biological Therapy, [online] 20(10), pp.1121–1124. doi:https://doi.org/10.1080/14712598.2020.1792880.
McColgan, P. and Tabrizi, S.J. (2017). Huntington’s disease: a clinical review. European Journal of Neurology, [online] 25(1), pp.24–34. doi:https://doi.org/10.1111/ene.13413.
uniQure (n.d.). AMT-130 Aims at the Core of Huntington’s Disease. Huntington’s Disease Association. Available at: https://www.hda.org.uk/seecmsfile/?id=239 [Accessed 5 Jan. 2026].
uniQure N.V. (2025) ‘uniQure announces positive topline results from pivotal Phase I/II study of AMT-130 in patients with Huntington’s disease’ [Press release], 24 September. Available at: https://uniqure.gcs-web.com/news-releases/news-release-details/uniqure-announces-positive-topline-results-pivotal-phase-iii (Accessed: 5 December 2025).